
Not just peer scientists, but even the general media like the New York Times is recognizing the AI team DeepMind for their incredible breakthrough results in the so-called “protein folding problem” and talking about how “this is a big deal”.
However, why is predicting the protein’s 3D structure so important? Also, how difficult can that be?
Through this article, I hope to convince you about the huge biological impact of AlphaFold advances on determining protein structures from its amino acid sequence and why the scientific community is so excited about it.
“In the drama of life on a molecular scale, proteins are where the action is”
Lesk, A. M., Introduction to Protein Architecture
Probably you have heard that proteins are considered fundamental for the structure and function of all organisms and for that reason they are usually described as the “building blocks of life”. They do most of the work in the cell; form important structures, transmit signals to regulate body tissues and organs, transport other molecules, carry out chemical reactions and even protect the body from bacterias and viruses. All processes taking place in an organism have proteins acting somewhere.
Why is Protein's 3D Structure so Important?
The human body contains a subset of 20,000 - 25,000 protein-coding genes with the ability to generate 20,000 to over millions of unique types of proteins.
Why so many?
Each has characteristic shapes, a 3D structure, that allow them to perform a precise function. As mentioned above, some proteins are structural, others transport other molecules, others are receptors, etc. The specific shape of each protein is tightly related to their function. For example, some proteins form pockets named active sites that perfectly fit to bind a particular target molecule.
The distinct “functional native structure” of proteins is important because it exposes several binding sites, channels, receptors and thus impacts how they bind other molecules or how proteins physically interact with others and assemble into complexes for structural or regulatory processes.
However, how do proteins reach their final form? Basically, they fold until acquiring their functional 3D native structure.
Every protein consists of a linear chain of basic units called amino acids, in average a protein is synthesized from 300 amino acids. The linear sequence of amino acids within a protein is considered the primary 1D structure and their ordering determines how the protein chain will fold up itself.
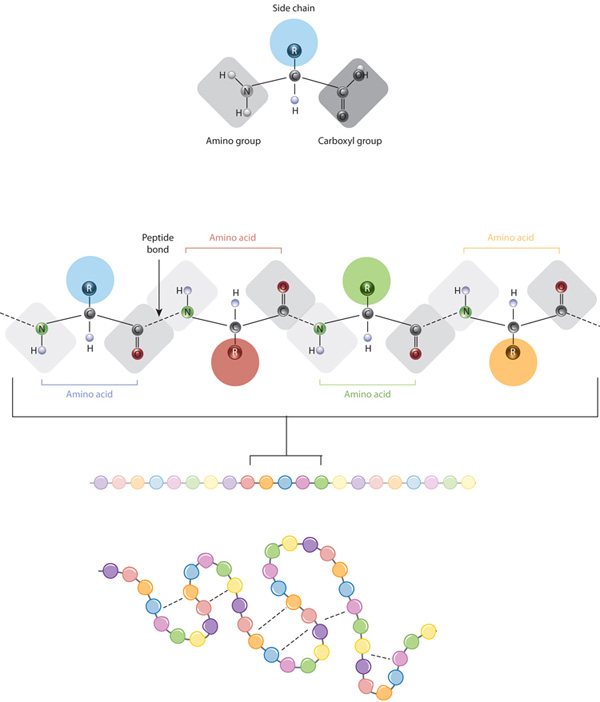
Figure 1: The relationship between amino acid side chains and protein conformation. The protein folds into a specific conformation depending on the interactions (dashed lines) between its amino acid side chains.
© 2010 Nature Education All rights reserved.
What Determines Protein Structure?
I just explained that the “recipe” proteins use to fold is in the sequence of amino acids, more specifically, protein folding is determined by the physicochemical properties that are encoded in the amino acids.
There is a set of 20 amino acids that made up proteins and all consist of an alpha (central) carbon atom linked to an amino (–NH2) and carboxyl (–COOH) functional groups, a hydrogen atom, and a variable side chain (R group) specific to each amino acid. The chemistry of these side chains is critical to protein structure. They can interact with other side chains driving the folding and intramolecular binding of the linear amino acid chain, which ultimately determine the protein’s shape or conformation (see Figure).
According to the laws of thermodynamics, systems gravitate toward their states of lower free energy, and the final shape adopted by proteins is also typically the most energetically favorable one. In the process of folding, a protein adopts a wide range of conformations before reaching their final, stable and unique form.
How Do Proteins Fold? Levinthal’s Paradox and the Protein Folding Problem
Now we know that the form of a protein is tightly related to its function. Knowledge of protein’s 3D structure is a huge hint for understanding how the protein works, and use that information for different purposes; control or modify protein’s function, predict what molecules bind to that protein and understand various biological interactions, assist drug discovery or even design our own proteins.
Yet, one of the biggest challenges of biology has been to accurately predict the 3D native structure of the protein from only its 1D sequence of amino acidic residues. Why is this a big problem?
The protein folding problem is stated in Levinthal’s paradox:
“Finding the native folded state of a protein by a random search among all possible configurations can take an enormously long time. Yet proteins can fold in seconds or less.”
citation: Zwanzig, R et al. “Levinthal's paradox"
From a general physicochemical point of view, how can proteins adopt their unique 3D native structure -a global free energy minimum form- in a biologically reasonable time without exhaustive enumeration of all possible conformations? This is under the assumption that proteins should randomly search configurations until the native form is reached.
Levinthal believed that proteins must solve the problem by folding through predetermined pathways.
DeepMind’s AI Program Solution to the Protein Folding Problem
Over the decades structural biologists have experimentally determined protein structures through complex methodologies (i.e. X-ray crystallography, nuclear magnetic resonance spectroscopy, electron microscopy and, in recent years, cryo-electron microscopy), but although they are very accurate, they cost millions and are extremely time-consuming. Hence, although our understanding has advanced considerably, it is still very limited to just a few protein structures. Thus, latest efforts have been placed in the prediction of protein 3D forms using computer-based algorithms.
Computer-based 3D structure prediction has been advanced by Professor John Moult and colleagues, in an event initiated in 1994 called CASP: Critical Assessment of protein Structure Prediction.
Google’s DeepMind participated for the first time in CASP13 in 2018, using deep-learning-based methods and won the competition. However, this year at CASP14 2020, the results of their AI program AlphaFold, have been praised by peer scientists. AlphaFold’s high accuracy to predict the 3D structure of proteins is believed to be of huge impact to life sciences and medicine.

AlphaFold predictions show high accuracy compared to experimental data.
Image from DeepMind's blog.
“It’s a game changer,” says Andrei Lupas, an evolutionary biologist at the Max Planck Institute for Developmental Biology in Tübingen, Germany. “This will change medicine. It will change research. It will change bioengineering. It will change everything,” Lupas added.
“This is a big deal” and “In some sense, the [protein folding] problem is solved”, commented professor Moult.
AlphaFold’s system develops strong predictions of the underlying physical structure of the protein, and also evaluates which predicted structures are reliable by using internal confidence measures.
In the competition, the predictions are given scores from 0 to 100 with 90 considered on par with the accuracy of experimental methods. AlphaFold’s predictions scored a median of 92.5 which is far better than its last score in CASP13. Some AlphaFold’s models were so good that in some cases defied the ones obtained by experimental approaches.
The Biological Impact and Future of AlphaFold
DeepMind’s system showed unprecedented results, but that neither means the protein folding problem is completely solved nor is the end of experimental methods to solve protein structure.
First, the system is trained on public dataset of known experimental protein structures, thus experimental approaches will still be required to contend with biases in the training data for the algorithm. For instance, there are many proteins with unknown 3D structures, therefore a bias could edge in toward those kinds of protein that have more structural data.
Additionally, although most of the predictions were highly accurate, the system is not perfect. The AI-based algorithm encountered difficulties modeling certain proteins, structures in protein complexes or in interaction with other proteins for instance.
What is important is that we only have experimental structures for just a small portion of all the different proteins that exist, and a system like AlphaFold will allow scientists to get a “good structure” using just publicly available and easier-to-collect experimental data.
Programs such as AlphaFold will exponentially increase our general understanding of different biological processes. As mentioned earlier, having a protein 3D structure is key to reveal the function of unknown proteins which would allow, for example, a better and accelerated understanding of diseases.
- Protein modeling methods could speed up drug development and reposition by predicting the effects of existing medication to new viruses. Pharma companies could generate their own antibody sequences in response to specific targets, use models for drug screening, and so on.
- It could also allow structure prediction for proteins that are not tenable for experimental determination, like very rare, scarce or ancient proteins. Model genetic variation into a protein structure for the evolutionary analysis of proteins.
- Last but not least, these tools could also be used for protein engineering. For example, we could design proteins with increased or reduced affinity to binding partners based on predicted structures. Furthermore, Briana Brownell, who is a data scientist at AI company PureStrategy, explained that we could potentially, “build proteins that fulfill a specific function.”She also added that “From protein therapeutics to biofuels or enzymes that eat plastic, the possibilities are endless.”
As a scientist myself I believe this is only the start of very exciting times for medicine and biotechnology, and can not wait for more "collaborative work" between artificial intelligence and other scientific fields.
What kind of problems will AI try to solve next?

![AlphaFold 2 and Related Patents: When AI Challenges the Protein Folding Problem [2]](https://ipms-content.piip.co.kr/article-images/1617930477157/5fcafea6-8c86-4c82-b987-e52357afd374/AlphaFold 2 and Related Patents.jpg)
![AlphaFold Breakthrough: When AI Challenges the Protein Folding Problem [1]](https://ipms-content.piip.co.kr/article-images/1617863166450/13d9777a-98ad-4e43-a045-b1870ee59419/predicting protein structure with AI.jpg)