
Late in 2020, DeepMind announced its AlphaFold 2 system and conducted major groundbreaking work using machine learning methods to predict the structure of proteins. The company, a subsidiary of Google-parent Alphabet, made the entire AlphaFold code available under the Apache License 2.0, and the model parameters are made available under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) license.
To date, many research groups have been taking advantage of the system as soon as the code was available. Some applications and optimizations include refined versions for the prediction of protein complexes, or others to speed up the predictions and make AF a more accessible platform. It is sure that AF2 and AlphaFold Protein Structure Database have opened the door for numerous applications and we will see many filings related to this technology.
Before, we discussed what was new in AlphaFold 2, but at the time we wrote our column, there was limited information and the AlphaFold article was not published. Still, people in the community made pretty good guessings about the main improvements of the system, and thus when the paper was finally released, there were not many surprises.
In resume,
AlphaFold 2 uses a graph network in combination with attention instead of convolutions. In AlphaFold 2 graph network, each amino acid is a node, and residues in close proximity define the edges to show how near one to another are the residue pairs. The system uses an attention-based model trained end-to-end to interpret the structure of the graph along with embed MSAs and amino acid residue pair representation to achieve iterative refinement of the amino acid graph representation from which 3D coordinates of all atoms of a protein are generated.
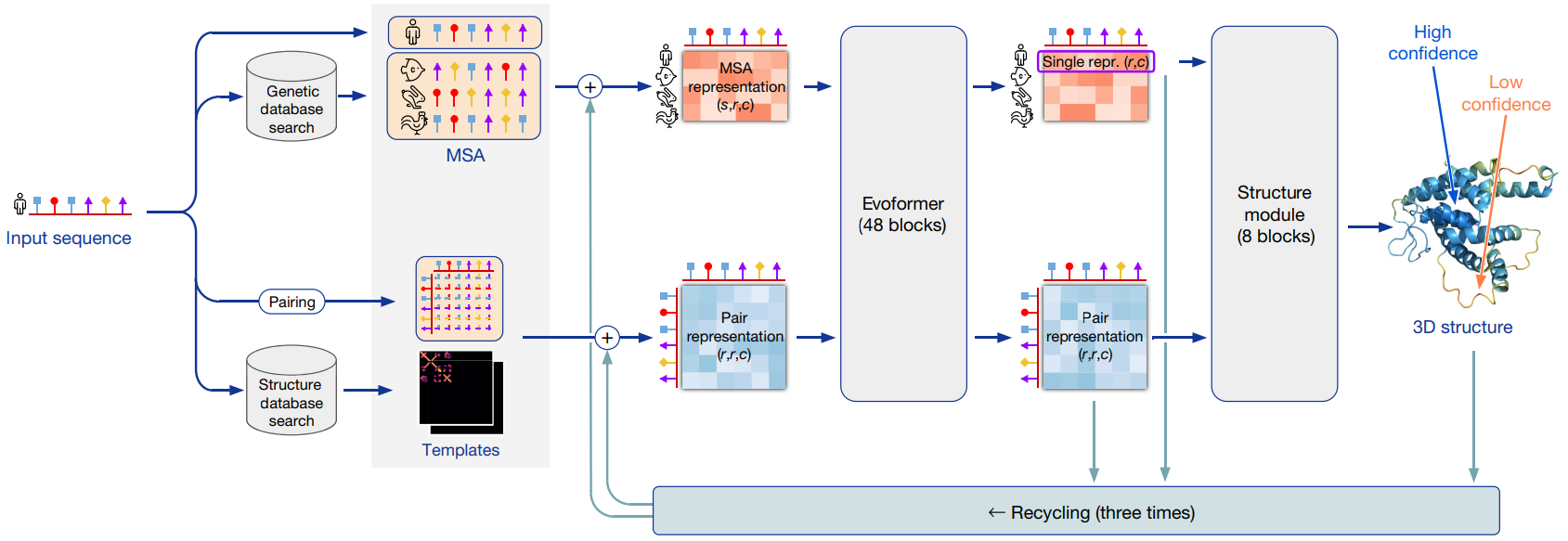
AlphaFold Overall Architecture and Improvements
Overall, AlphaFold 2 (AF2) system can be divided into three main parts. In the first step, 1) the system embeds MSA information corresponding to the input sequence, sequence information, and template information into the MSA representation and pair representation. Then 2) updates the two representations using the “Evoformer”, which is composed of two transformers. And 3) the “Structure Module” estimates a 3D structure based on the last updated output representations from the Evoformer. Additionally, in AF2, the results of the entire process are sent backward or “recycled”, so the MSA and pair representations are in constant talk that keeps updating and refining both representations.
Next, let’s examine some of the steps and the improvements of AlphaFold 2 in comparison with its previous version.
1. Embeddings for the MSA and templates: Starting from the input sequence, AF searches for similar sequences using MSAs. An important difference is that AF1 used only MSA statistics, while AF2 embeds raw sequences found by MSA.
This is relevant in the context of the protein structure problem in which one would like to take advantage of the information from sequences with unknown unlabeled structures. Via embeddings, AF2 can obtain a more complex understanding of the correlation between amino acids. Even though we don’t know the structure of all the sequences, the correlated mutations from the MSAs are informative.
How it works: In the embedding process, in order to reduce the loaded MSA information, AF2 groups all similar sequences by similarity (cluster), and a representative is chosen from each cluster to form the MSA (msa_feat). These representatives are embedded with all features that describe the cluster, so all sequences influence the final prediction and there is no significant loss of accuracy.
Additionally to the MSAs, AF2 also embeds a pair representation, representing the distance between residues. This may be comparable to the distogram presented in AF1. The pair representation is updated with the input sequence information and the extra MSA presentation embedding (extra_msa_feat) that correspond to non-clustered MSA information.
2. Graph and attention over convolutions: AlphaFold 1 is defined as a convolutional neural network (CNN) trained on known protein structures and features derived from evolutionarily related sequences (MSA) to predict the distance between pairs of amino acids of an input sequence in order to generate a 3D structure. But CNNs in AF1 might not take advantage of all the information contained in the sequences since they tend to ignore long-range dependencies between residues, which in many cases are important to protein shape. When detecting objects in a picture, the detection of an object is not affected by something in the corner of the image, so CNNs are probed to be useful. But they may not be the best option in problems where the positioning in the sequence conveys a meaning, such as the protein folding problem. Attention mechanisms, on the other hand, are supposed to perceive those important elements.
An essential component of AlphaFold 2 is the Evoformer, which uses attention-based mechanisms that allow it to learn correlations in the input data. This attention mechanism gives more flexibility to the network, allowing it to dynamically learn interactions between non-neighboring nodes. In this manner, the system can learn what relations are relevant and which can be ignored to focus on computing the graph of how each residue relates to another.

How it works: The evoformer consists of 48 blocks, mostly composed of attention-based layers, that refines both; the MSA and pair representations. There are also clear communication channels between the two sections that update both representations.
When dealing with the pair representation, the information is transformed and arranged into graphs, and iteratively refined in the form of triangular updates.
In the section that deals with the MSA representation, the network first computes row-wise attention (horizontal direction), identifying which amino acids in the sequence are more related to each other. When updating the relation between amino acids, the network adds a pair bias term that is calculated directly from the current pair representation. By using this bias value, it helps the network to augment the attention mechanisms and find more accurately the relationship between amino acids.

Then, column-wise attention (vertical direction) to identify which sequences are more important. It exchanges information between sequences within an alignment column and helps the network to propagate information on 3D structure from the first sequence of the alignment (which is the sequence for which we predict the structure in the next step) to the others.
Two points of communication to refine the updates can be found in the evoformer section. The first we already mentioned, is the bias in where the pair representation communicates and helps update the MSA. The outer product mean block is the other communication point, where the MSA representation provides a way to update the pair representation.
As mentioned before, when refining the pair representation, AF2 uses attention to process how residue pairs relate to one another in a graph representation. This graph representation is arranged in terms of a triplet of amino acids (nodes) and their corresponding edges.

Enforces the idea of using the triangle inequality for the pair distances distribution to be representable as a 3D structure. On this basis, the pair representation is being updated in a process called triangle multiplicative update that updates an edge (distance between residues) based on the two other nodes and their edges.
Then these three nodes are updated using triangular self-attention. Attention allows further geometrical and chemical constraints to be learned by the algorithm while triangular multiplicative updates ensure that no excess attention is paid to a particular residue subsequence. The idea of using these triangles and incorporating the “missing” connecting edge informs the position of the residues with respect to global attractions. For example, between partially charged residues that may be very far away in the sequence but are near in the 3D form.
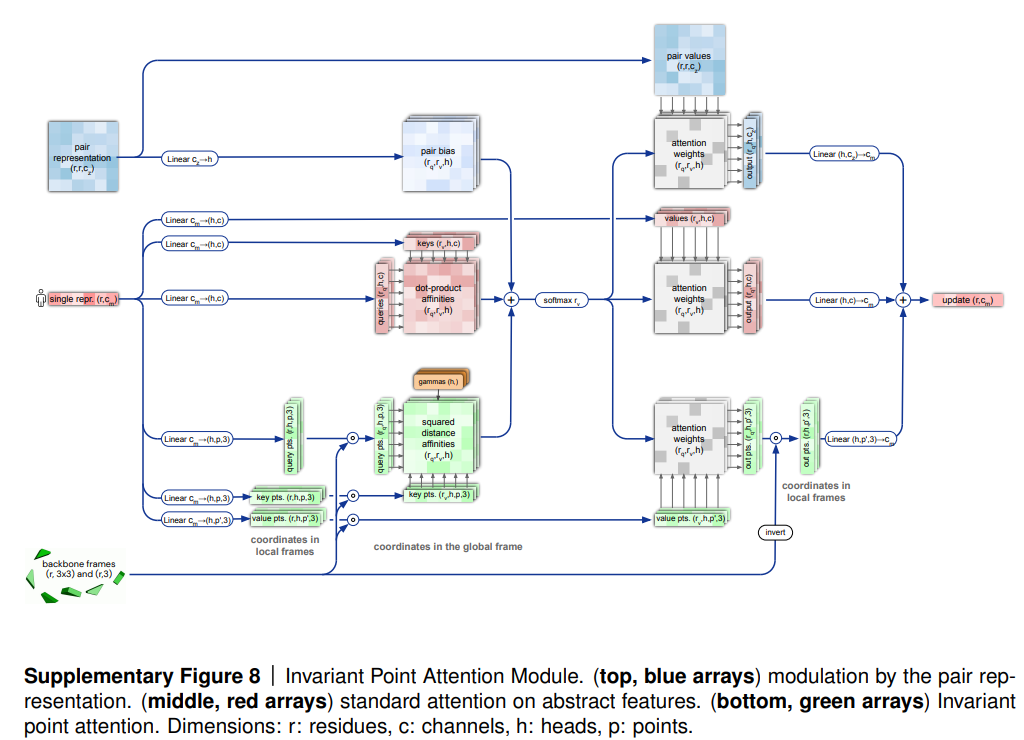
3. Adaptative Computation Time: The idea of adaptive computation time (ACT) refers to a mechanism that allows an RNN to dynamically learn how many repetitions need to evaluate its input before outputting it to the next step. Deep networks may perform better, but it could be optimal if the network could take a cost-benefit decision on how deep it should go to be more computationally efficient. In the case of AF2, ACT is achieved -at least partially- through invariant point attention.
How it works: The Invariant Point Attention (IPA) module models rotations and displacing of each independent amino acid (as triangles in space) to produce a final 3D structure.
IPA is a new type of attention devised specifically for working with 3D. By embedding the equivariance property in the model, the network understands that rotating the global frame is inconsequential to the predictions. Since IPA frees the network from having to learn equivariance, it concentrates on maximal extraction of information in each cycle of training, and in consequence it reduces the number of models the network asses to find the correct folding.

4. End-to-End Prediction: Jumper and colleagues have emphasized training AlphaFold 2 from end-to-end, so it is constantly refining its models of the protein. The system continually feeds all the outputs from the evoformer, and the structure model (i.e. MSA and pair representations and predicted structure) back into the start of the process until it can no longer improve. This iterative refinement within the whole network (or ‘recycling’) contributes to eliminating the disconnection between pairwise distance predictions and the 3D structure encountered in the previous AlphaFold.
Alpha Fold 2 tool and Alpha Fold Protein Database are for sure catalyzing new discoveries in the industry, especially in pharma, by enabling faster and more accurate protein predictions
We have discussed some of the features of AlphaFold 2 and some points we consider to be the main improvements and ideas in comparison with other tools and its own previous version. But what is more remarkable is the fact that all features that contributed to AF2 sophisticated architecture were introduced in other networks, and in the same manner, they have the potential to be applicable to other systems for solving diverse problems.
Hopefully, in a near future, we will witness more advanced AI tools based on the presented ideas that could accomplish even more outstanding results.
References:
- AlphaFold 2 is here: what’s behind the structure prediction miracle
- How DeepMind AlphaFold2 works?
